In this subgroup of the CWI Life Sciences group, led by Prof. dr. Leen Stougie, we are currently working on two separate topics, metabolic networks and phylogeny/phylogenetic networks. In the past we have also worked on problems related to haplotyping and genome reversals. Our subgroup has a strong focus on combinatorial optimization and approximation algorithms.
1. Metabolic networks
In constraint-based models of metabolic networks it is common to model the system as Nx=0, where N is a stochiometric matrix. Rows of N represent metabolites and columns of N represent reactions. The idea of using the null space is that (within appropriate time intervals) production and consumption of metabolites are assumed balanced. Often some entries of the x vector are constrained to be non-negative. Biologists are generally interested in support-minimal solutions to the system, known as Elementary Flux Modes (EFMs). Computation of EFMs is closely related to enumeration of the extreme rays of a polyhedral cone. We study problems related to EFM generation and other problems that have their roots in pathways through metabolic networks, such as precursor set enumeration.
Often there are additional environmental constraints resulting from Flux Balance Analysis (FBA) experiments. The system becomes a polyhedron, rather than a cone. This FBA-polyhedron has interesting properties, which we are currently studying.
Our work on metabolic networks ainly takes place within two separate projects:
- MEMESA - Microbial Ecosystems and Multiple Environment Stochiometric Analyses
- Research within the INRIA Associated Team Project SIMBIOSI
See also the webpage of Leen Stougie at the Vrije Universiteit (Amsterdam).
2. Phylogeny / Phylogenetic Networks
The traditional model for representing evolution is the phylogenetic tree. The classic example of this is the Tree of Life. This is the idea popularised by Charles Darwin that all life on Earth is descended from a single ancient common ancestor and that some species are more related than others. For example, humans and apes are clearly closer relatives than humans and fish. The phylogenetic tree allows us to model evolutionary events such as mutation and speciation (when one species splits up into two or more subspecies). The classical problem of phylogenetics is to take a set of species and infer (by looking at their DNA for example) the ''most likely'' tree explaining their evolution. This problem is in general difficult, but has been extremely well studied.
In recent years, however, awareness has grown of reticulate evolutionary phenomena. These concern evolutionary events where a species does not split into two or more subspecies, but where two or more already existing species fuse together to form a new species. Hybridization and Horizontal Gene Transfer (HGT) are examples of these. Phylogenetic trees cannot model these events well. It is much more natural to model reticulate events as vertices with indegree 2 or higher. Hence the use of phylogenetic networks, essentially a generalisation of trees to directed acyclic graphs (DAGs). This leads to new challenges both from a modelling and optimization/efficiency perspective. Our group attempts to tackle these challenges using tools from combinatorial optimization and efficient (approximation) algorithm design.
See the website of Steven Kelk for a list of publications and software packages.
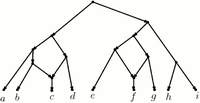
A level-1 phylogenetic network